Sanaris's answer is a great, succinct list of what each term in the free energy expression stands for: I'm going to concentrate on the $T\,S$ term (which you likely find the most mysterious) and hopefully give a little more physical intuition. Let's also think of a chemical or other reaction, so that we can concretely talk about a system changing and thus making some of its internal energy $H=U+p\,V$ available for work.
The $T S$ term arises roughly from the energy that is needed to "fill up" the rotational, vibrational, translational and otherwise distractional thermal energies of the constituents of a system. Simplistically, you can kind of think of its being related to the idea that you must use some of the energy released to make sure that the reaction products are "filled up" with heat so that they are at the same temperature as the reactants. So the $T S$ term is related to, but not the same as, the notion of heat capacity: let's look at this a bit further.
Why can't we get at all the energy $\Delta H$? Well, actually we can in certain contrived circumstances. It's just that these circumstances are not useful for calculating how much energy we can practically get to. Let's think of the burning of hydrogen:
$$\rm H_2+\frac{1}{2} O_2\to H_2O;\quad\Delta H \approx 143{\rm kJ\,mol^{-1}}\tag{1}$$
This is a highly exothermic one, and also one of the reactions of choice if you want to throw three astronauts, fifty tons of kit and about a twentieth of the most advanced-economy-in-the-world’s-1960s GDP at the Moon.
The thing about one mole of $H_2O$ is that it can soak up less heat than the mole of $H_2$ and half a mole of $O_2$; naively this would seem to say that we can get more heat than the enthalpy change $\Delta H$, but this is not so. We imagine a thought experiment, where we have a gigantic array of enormous heat pads (all individually like an equilibrium “outside world") representing all temperatures between absolute zero and $T_0$ with a very fine temperature spacing $\Delta T$ between them. On my darker days I find myself imagining an experimental kit that looks eerily like a huge pallet on wheels of mortuary shelves, sliding in and out as needed by the experimenter! We bring the reactants into contact with the first heat pad, which is at a temperature $T_1 = T_0 - \Delta T$ a teeny-tiny bit cooler than $T_0$ thus reversibly drawing some heat $\Delta Q(T_1)$ off into the heat pad. Next, we bring the reactants into contact with the second heat pad at temperature $T_2 = T_0 - 2\,\Delta T$, thus reversibly drawing heat $\Delta Q(T_2)$ off into that heat pad. We keep cooling then shifting to the next lowest heat pad until we have visited all the heat pads and thus sucked all the heat off into our heat pads: see my sketch below:
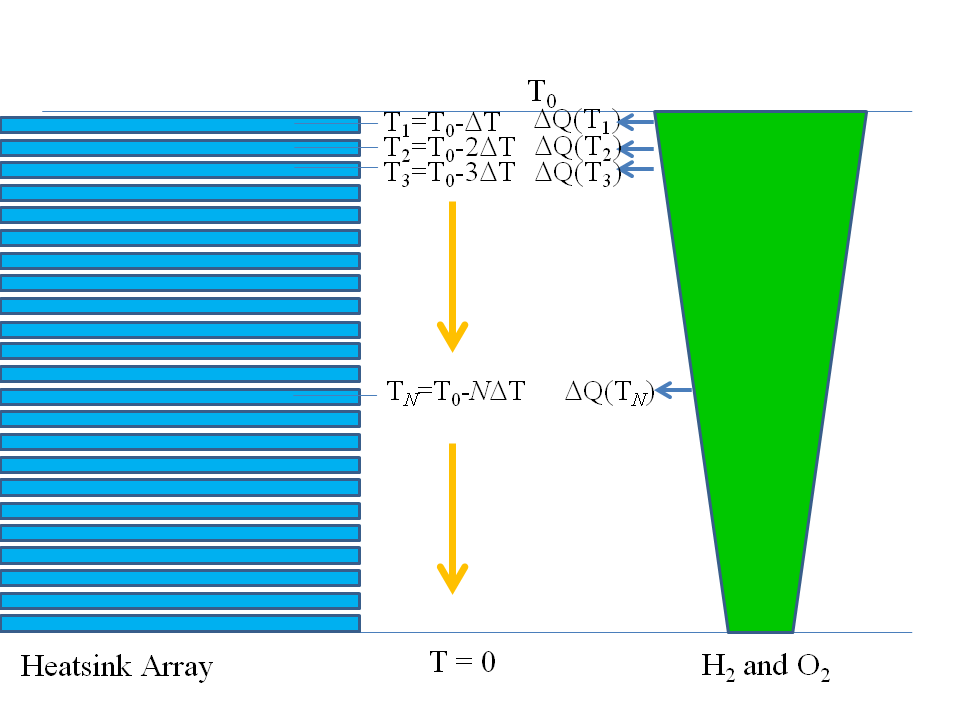
Now the reactants are at absolute zero. There is no heat needed to "fill them up" to their temperature, so we can extract all the enthalpy $\Delta H$ from the reaction as useful work. Let's imagine we can put this work aside in some ultrafuturistic perfect capacitor, or some such lossless storage for the time being.
Now we must heat our reaction products back up to standard temperature, so that we know what we can get out of our reaction if the conditions do not change. So, we simply do the reverse, as sketched below:
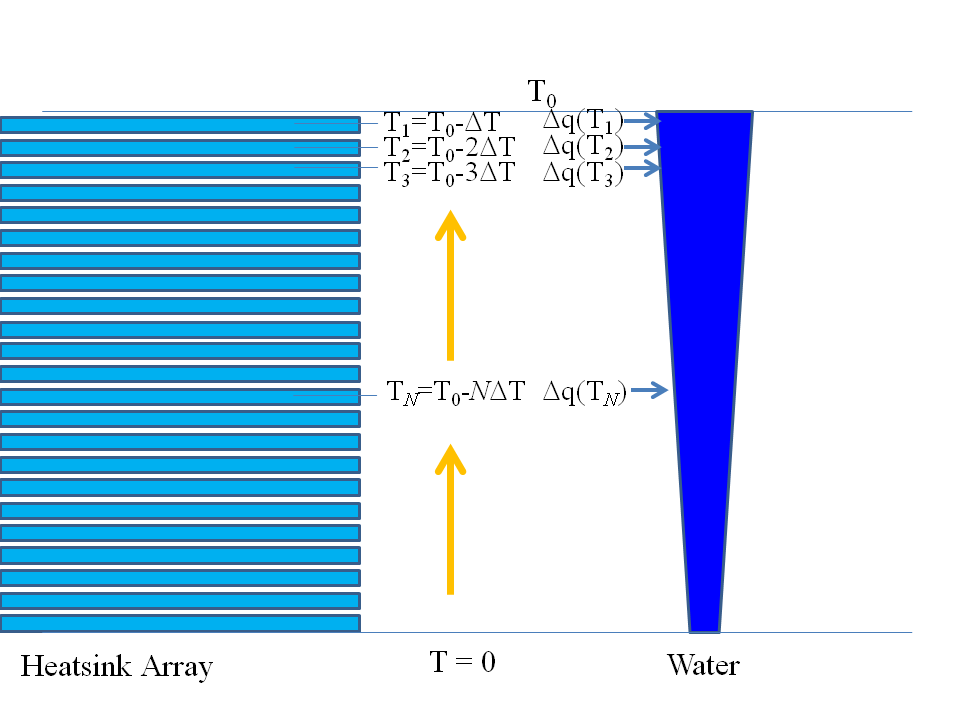
Notice that I said that $H_2O$ soaks up less heat than the reactants. This means that, as we heat the products back up to standard temperature, we take from the heat pads less heat in warming up the water than we put into them in cooling the reactants down.
So far, so good. We have gotten all the work $\Delta H$ out into our ultracapacitor without losing any! And we're back to our beginning conditions, or so it seems! What's the catch?
The experimental apparatus that let us pull this trick off is NOT back at its beginning state. We have left heat in the heat pads. We have thus degraded them: they have warmed up ever so slightly and so cannot be used indefinitely to repeatedly do this trick. If we tried to do the trick too many times, eventually the heat pads would be at ambient temperature and would not work any more.
So we haven’t reckoned the free energy at the standard conditions, rather we have simply calculated the free energy $\Delta H$ available in the presence of our unrealistic heat sink array. To restore the system to its beginning state and calculate what work we could get if there were no heat sink array here, we must take away the nett heat flows we added to all the heat pads and send them into the outside World at temperature $T_0$. This is the only "fair" measure, because it represents something that we could do with arbitrarily large quantities of reactants.
But the outside World at $T_0$ is warmer than any of the heat pads, so of course this heat transfer can’t happen spontaneously, simply by dent of Carnot’s statement of the second law!
We must therefore bring in a reversible heat pump and use some of our work $\Delta H$ to pump this heat into the outside world to restore standard conditions: we would connect an ideal reversible heat pump to each of the heat pads in turn and restore them to their beginning conditions, as sketched below:
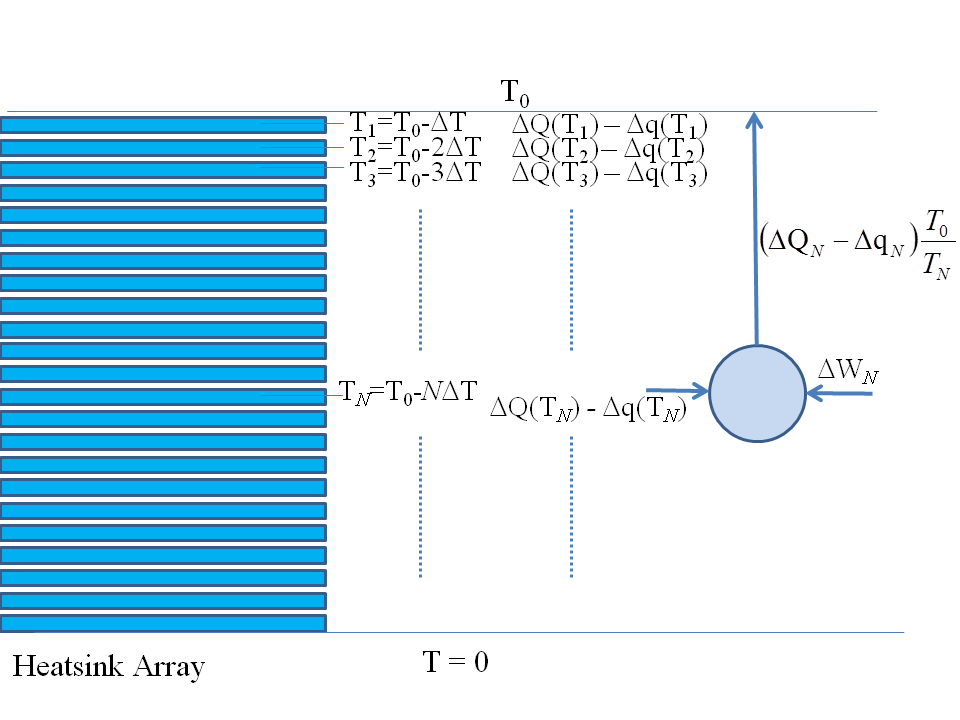
This part of the work that we use to run the heat pump and restore all the heat pads, if you do all the maths, is exactly the $T\,\Delta S$ term.
The above is a mechanism whereby the following statement in Jabirali's Answer holds:
Processes that increase the Gibbs free energy can be shown to increase the entropy of the system plus its surroundings, and will therefore be prevented by the second law of thermodynamics.
The nice thing about the above is that it is a great way to look at endothermic reactions. In an endothermic reaction, we imagine having an energy bank that we can borrow from temporarily. After we have brought the products back up to temperature, we find we have both borrowed $-\Delta H$ from the energy bank and put less heat back into the heat pads than we took from them. So heat can now flow spontaneously from the environment to the heat pads to restore their beginning state, because the heat pads are all at a lower temperature than the environment. As this heat flows, we can use a reversible heat engine to extract work from the heat flowing down the gradient. This work is, again, $-T\,\Delta S$, which is a positive work gotten from the heat flowing down the temperature gradient. The $-T\,\Delta S$ can be so positive that we can pay back the $\Delta H$ we borrowed and have some left over. If so, we have an endothermic reaction, and a nett free energy: this energy coming from the heat flowing spontaneously inwards from the environment to fill the higher entropy products (higher than the entropy of the reactants).
Take heed that, in the above, I have implicitly assumed the Nernst Heat Postulate -the not quite correct third law of thermodynamics - see my answer here for more details. For the present discussion, this approximate law is well good enough.