The Brillouin Zone (BZ) refers to a region of reciprocal space corresponding to the primitive cell. That is, a Brillouin Zone is a subset of the reciprocal space which contains all the information necessary to describe the crystal.
In the case of a crystal which only has a single type of atom, the procedure for determining the corresponding BZ is simple:
- Take the lattice in the real space and convert it to the recirpocal space


Choose any one lattice site as the origin and draw lines connecting it to all of its nearest neighbours
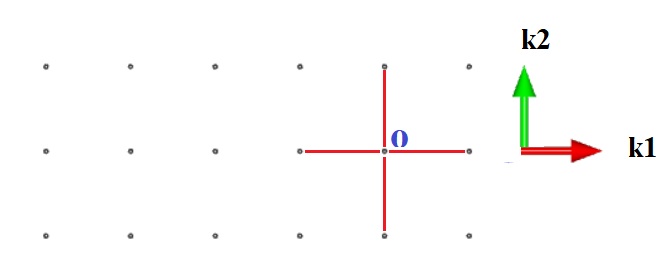
Draw the perpendicular bisectors of these connecting lines. The area bounded by the bisectors is the first Brillouin Zone

Wikipedia has a very clear illustration of the process.
{kind=link}
It is further possible to find the Irreducible part of the Brillouin Zone (IBZ), which is the smallest possible subset of the BZ after reducing it along its symmetries in the same ways as the symmetries present in the point group associated with the lattice.

The Problem:
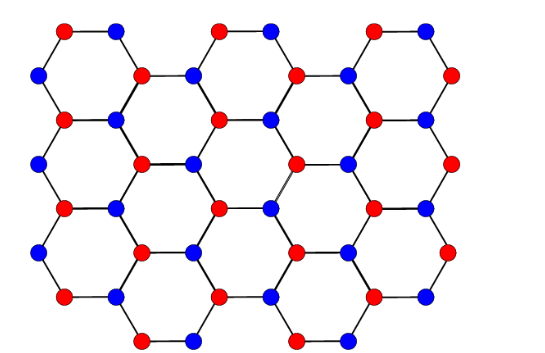
The above formulation is clear and well-known. However, I am not very sure about what to do if the crystal has more than one atom. There can be several possibilities:
- We could treat the question as purely concerning the lattice and treat all the atoms as if they were identical. Then follow the earlier process.
- We could treat the crystal as having a motif consisting of groups of the different atoms. Then, try to obtain a resulting lattice and carry out the standard process on it.
- Connect nearest neighbours using sets of either only identical or only dissimilar atoms and continue the earlier process.
Unfortunately, none of these options seems obviously correct. It feels like there should be a clear answer based on the underlying theory, but I have had no luck finding it so far.
What is the correct way to find the Brillouin Zone/ Irreducible Brillouin Zone for a crystal cconsisting of more than one type of atom? (such as the one shown below)